
Paper List
-
Nyxus: A Next Generation Image Feature Extraction Library for the Big Data and AI Era
This paper addresses the core pain point of efficiently extracting standardized, comparable features from massive (terabyte to petabyte-scale) biomedi...
-
Topological Enhancement of Protein Kinetic Stability
This work addresses the long-standing puzzle of why knotted proteins exist by demonstrating that deep knots provide a functional advantage through enh...
-
A Multi-Label Temporal Convolutional Framework for Transcription Factor Binding Characterization
This paper addresses the critical limitation of existing TF binding prediction methods that treat transcription factors as independent entities, faili...
-
Social Distancing Equilibria in Games under Conventional SI Dynamics
This paper solves the core problem of proving the existence and uniqueness of Nash equilibria in finite-duration SI epidemic games, showing they are a...
-
Binding Free Energies without Alchemy
This paper addresses the core bottleneck of computational expense in Absolute Binding Free Energy calculations by eliminating the need for numerous al...
-
SHREC: A Spectral Embedding-Based Approach for Ab-Initio Reconstruction of Helical Molecules
This paper addresses the core bottleneck in cryo-EM helical reconstruction: eliminating the dependency on accurate initial symmetry parameter estimati...
-
Budget-Sensitive Discovery Scoring: A Formally Verified Framework for Evaluating AI-Guided Scientific Selection
This paper addresses the critical gap in evaluating AI-guided scientific selection strategies under realistic budget constraints, where existing metri...
-
Probabilistic Joint and Individual Variation Explained (ProJIVE) for Data Integration
This paper addresses the core challenge of accurately decomposing shared (joint) and dataset-specific (individual) sources of variation in multi-modal...
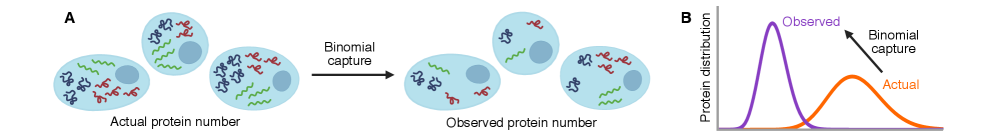
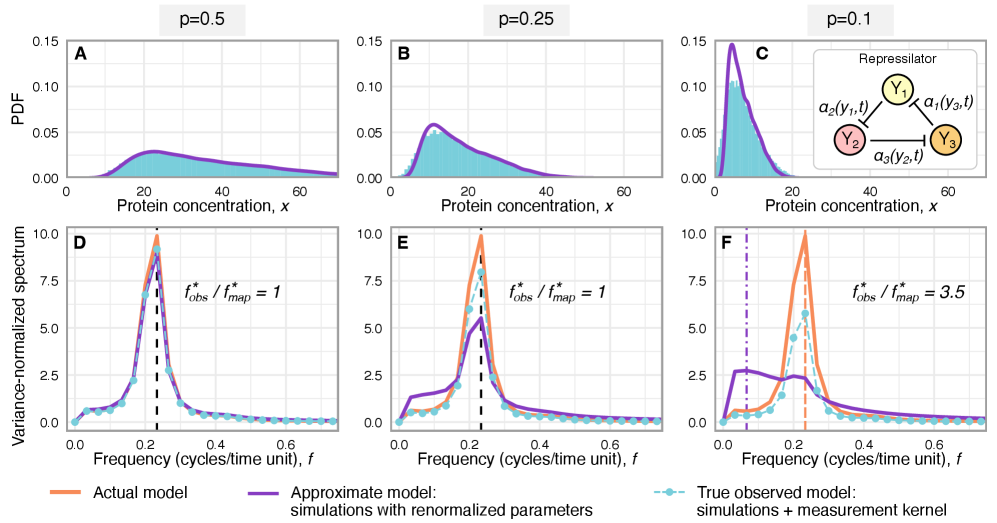
Imperfect molecular detection renormalizes apparent kinetic rates in stochastic gene regulatory networks
Department of Mathematical Analysis and Numerical Mathematics, Comenius University, Slovakia | University of Edinburgh, UK
30秒速读
IN SHORT: This paper addresses the core challenge of distinguishing genuine stochastic dynamics of gene regulatory networks from artifacts introduced by imperfect molecular detection in single-cell experiments.
核心创新
- Methodology Extends the binomial capture model from simple gene expression to general gene regulatory networks (GRNs) with explicit regulation, enabling analysis of technical noise in complex systems.
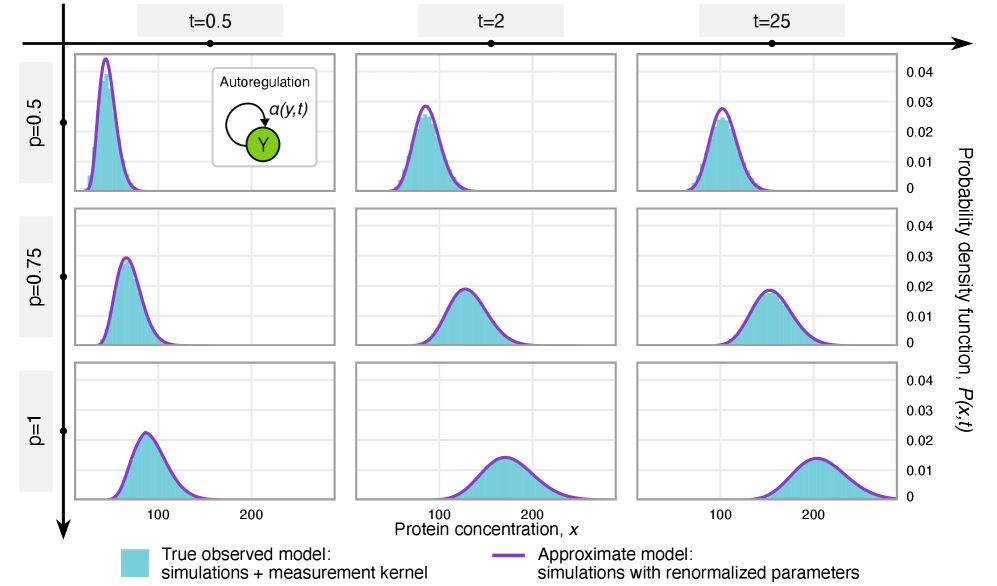
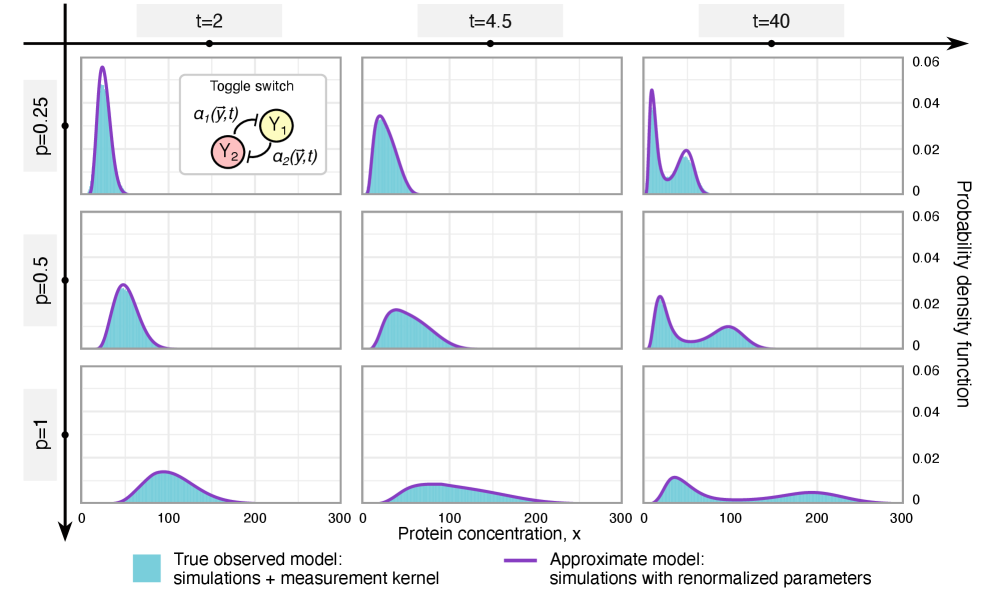
- Theory Establishes precise mathematical conditions under which technical noise leads to a renormalization (rescaling) of kinetic rates versus when it introduces non-absorbable distortions.
- Methodology Derives results valid for networks of arbitrary connectivity and under time-dependent kinetic rates, significantly generalizing previous steady-state analyses.
主要结论
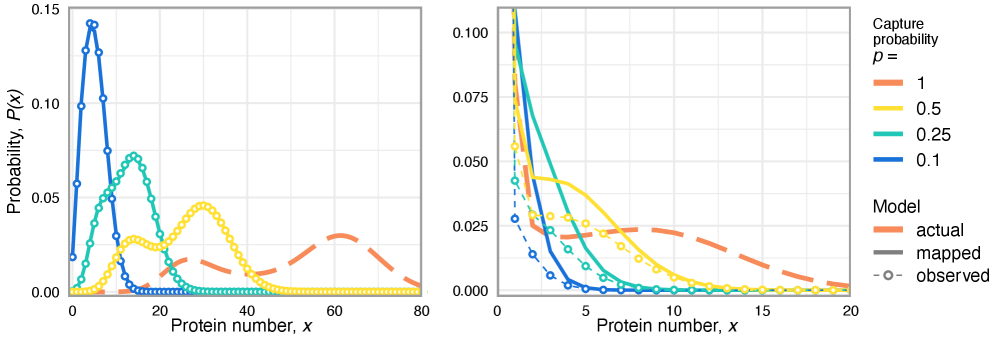
- Technical noise systematically reduces the apparent mean burst size of gene products by a factor of p (the capture probability), e.g., from b(t) to b(t)*p.
- Rate renormalization occurs when promoter-state transitions are on a distinct timescale (much slower/faster) than other reactions or under high transcription factor abundance.
- The framework shows that for the telegraph model, the observed mRNA dynamics are equivalent to the true system with a renormalized transcription rate: k₃(t) → p*k₃(t).
摘要: Imperfect molecular detection in single-cell experiments introduces technical noise that obscures the true stochastic dynamics of gene regulatory networks. While binomial models of molecular capture provide a principled description of imperfect detection, they have so far been analyzed only for simple gene-expression models that do not explicitly account for regulation. Here, we extend binomial models of capture to general gene regulatory networks to understand how imperfect capture reshapes the observed time-dependent statistics of molecular counts. Our results reveal when capture effects correspond to a renormalization of a subset of the kinetic rates and when they cannot be absorbed into effective rates, providing a systematic basis for interpreting noisy single-cell measurements. In particular, we show that rate renormalization emerges either under significant transcription factor abundance or when promoter-state transitions occur on a distinct (much slower or faster) timescale than other reactions. In these cases, technical noise causes the apparent mean burst size of synthesized gene products to appear reduced while transcription factor binding reactions appear faster. These effects hold for gene regulatory networks of arbitrary connectivity and remain valid under time-dependent kinetic rates.