
Paper List
-
Discovery of a Hematopoietic Manifold in scGPT Yields a Method for Extracting Performant Algorithms from Biological Foundation Model Internals
This work addresses the core challenge of extracting reusable, interpretable, and high-performance biological algorithms from the opaque internal repr...
-
MS2MetGAN: Latent-space adversarial training for metabolite–spectrum matching in MS/MS database search
This paper addresses the critical bottleneck in metabolite identification: the generation of high-quality negative training samples that are structura...
-
Toward Robust, Reproducible, and Widely Accessible Intracranial Language Brain-Computer Interfaces: A Comprehensive Review of Neural Mechanisms, Hardware, Algorithms, Evaluation, Clinical Pathways and Future Directions
This review addresses the core challenge of fragmented and heterogeneous evidence that hinders the clinical translation of intracranial language BCIs,...
-
Less Is More in Chemotherapy of Breast Cancer
通过纳入细胞周期时滞和竞争项,解决了现有肿瘤-免疫模型的过度简化问题,以定量比较化疗方案。
-
Fold-CP: A Context Parallelism Framework for Biomolecular Modeling
This paper addresses the critical bottleneck of GPU memory limitations that restrict AlphaFold 3-like models to processing only a few thousand residue...
-
Open Biomedical Knowledge Graphs at Scale: Construction, Federation, and AI Agent Access with Samyama Graph Database
This paper addresses the core pain point of fragmented biomedical data by constructing and federating large-scale, open knowledge graphs to enable sea...
-
Predictive Analytics for Foot Ulcers Using Time-Series Temperature and Pressure Data
This paper addresses the critical need for continuous, real-time monitoring of diabetic foot health by developing an unsupervised anomaly detection fr...
-
Hypothesis-Based Particle Detection for Accurate Nanoparticle Counting and Digital Diagnostics
This paper addresses the core challenge of achieving accurate, interpretable, and training-free nanoparticle counting in digital diagnostic assays, wh...
Simulation and inference methods for non-Markovian stochastic biochemical reaction networks
School of Mathematical Sciences, Queensland University of Technology | Centre for Data Science, Queensland University of Technology | ARC Centre of Excellence for Mathematical Analysis of Cellular Systems (MACSYS), Queensland University of Technology
30秒速读
IN SHORT: This paper addresses the computational bottleneck of simulating and performing Bayesian inference for non-Markovian biochemical systems with history-dependent delays, which are crucial for modeling processes like gene transcription but are prohibitively expensive with existing methods.
核心创新
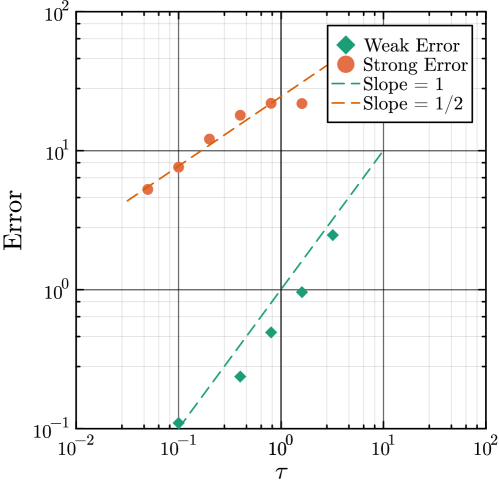
- Methodology Generalizes the next reaction method and τ-leaping to support arbitrary inter-event time distributions for non-Markovian systems, maintaining computational scalability.
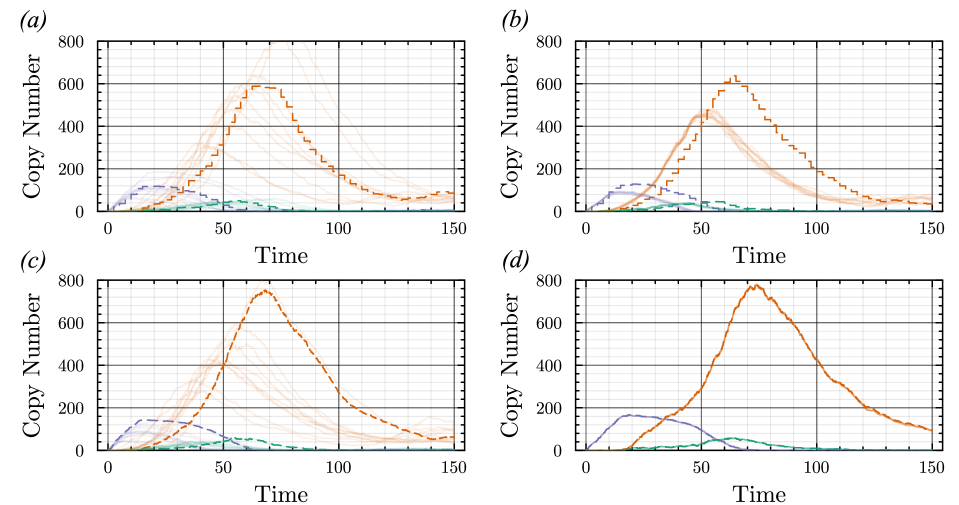
- Methodology Introduces a novel coupling scheme to generate positively correlated exact and approximate non-Markovian sample paths, a prerequisite for variance reduction techniques.
- Methodology Enables the application of multifidelity and multilevel Monte Carlo (MLMC) methods to non-Markovian systems for the first time, bridging a significant methodological gap.
主要结论
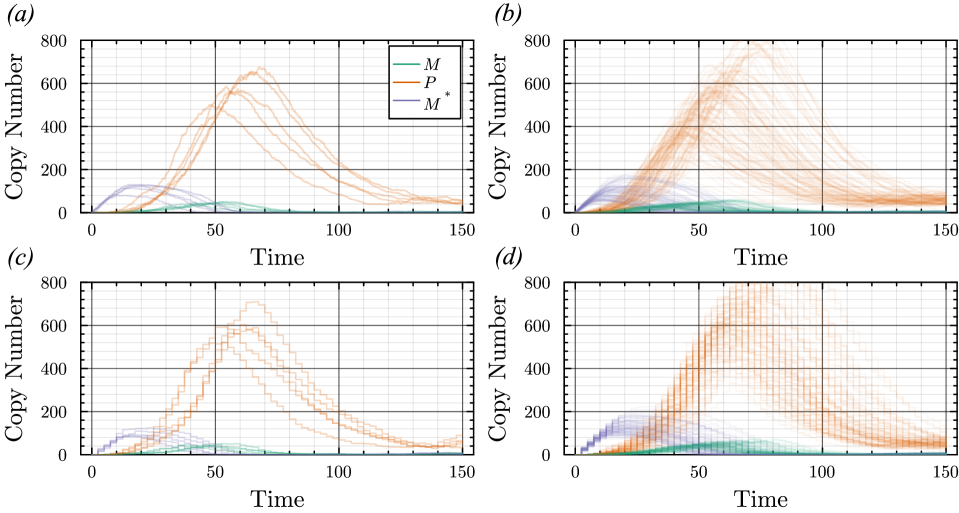
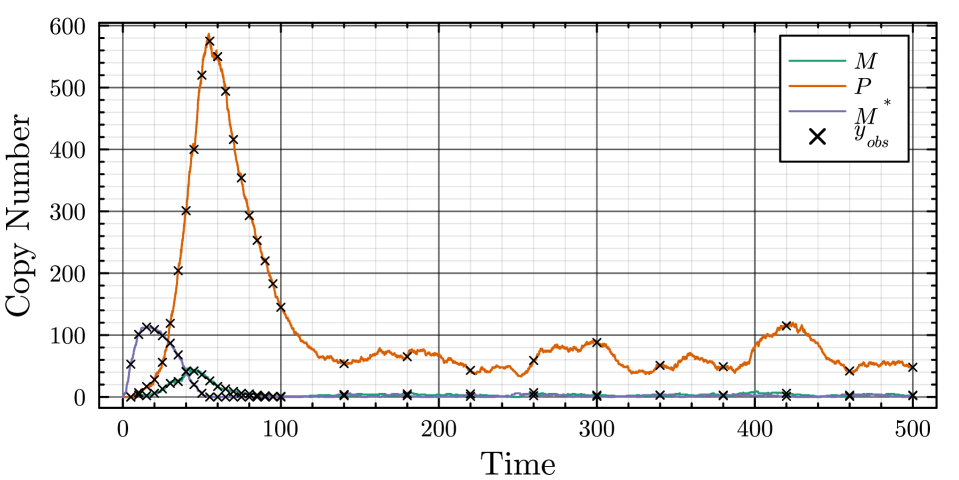
- The proposed non-Markovian simulation algorithms and coupling scheme successfully enable multifidelity inference, demonstrated on a gene regulation model with delayed auto-inhibition.
- The method achieves a computational speedup of two orders of magnitude (100x) in inference efficiency compared to standard approaches for the non-Markovian case study.
- The framework supports arbitrary delay distributions (state- and time-dependent), significantly extending the practical modeling scope beyond previous methods limited to simpler, time-only delays.
摘要: Stochastic models of biochemical reaction networks are widely used to capture intrinsic noise in cellular systems. The typical formulation of these models are based on Markov processes for which there is extensive research on efficient simulation and inference. However, there are biological processes, such as gene transcription and translation, that introduce history dependent dynamics requiring non-Markovian processes to accurately capture the stochastic dynamics of the system. This greater realism comes with additional computational challenges for simulation and parameter inference. We develop efficient stochastic simulation algorithms for well-mixed non-Markovian stochastic biochemical reaction networks with delays that depend on system state and time. Our methods generalize the next reaction method and τ-leaping method to support arbitrary inter-event time distributions while preserving computational scalability. We also introduce a coupling scheme to generate exact non-Markovian sample paths that are positively correlated to an approximate non-Markovian τ-leaping sample path. This enables substantial computational gains for Bayesian inference of model parameters though multifidelity simulation-based inference schemes. We demonstrate the effectiveness of our approach on a gene regulation model with delayed auto-inhibition, showing substantial gains in both simulation accuracy and inference efficiency of two orders of magnitude. These results extend the practical applicability of non-Markovian models in systems biology and beyond.