
Paper List
-
Ill-Conditioning in Dictionary-Based Dynamic-Equation Learning: A Systems Biology Case Study
This paper addresses the critical challenge of numerical ill-conditioning and multicollinearity in library-based sparse regression methods (e.g., SIND...
-
Hybrid eTFCE–GRF: Exact Cluster-Size Retrieval with Analytical pp-Values for Voxel-Based Morphometry
This paper addresses the computational bottleneck in voxel-based neuroimaging analysis by providing a method that delivers exact cluster-size retrieva...
-
abx_amr_simulator: A simulation environment for antibiotic prescribing policy optimization under antimicrobial resistance
This paper addresses the critical challenge of quantitatively evaluating antibiotic prescribing policies under realistic uncertainty and partial obser...
-
PesTwin: a biology-informed Digital Twin for enabling precision farming
This paper addresses the critical bottleneck in precision agriculture: the inability to accurately forecast pest outbreaks in real-time, leading to su...
-
Equivariant Asynchronous Diffusion: An Adaptive Denoising Schedule for Accelerated Molecular Conformation Generation
This paper addresses the core challenge of generating physically plausible 3D molecular structures by bridging the gap between autoregressive methods ...
-
Omics Data Discovery Agents
This paper addresses the core challenge of making published omics data computationally reusable by automating the extraction, quantification, and inte...
-
Single-cell directional sensing at ultra-low chemoattractant concentrations from extreme first-passage events
This work addresses the core challenge of how a cell can rapidly and accurately determine the direction of a chemoattractant source when the signal is...
-
SDSR: A Spectral Divide-and-Conquer Approach for Species Tree Reconstruction
This paper addresses the computational bottleneck in reconstructing species trees from thousands of species and multiple genes by introducing a scalab...
Simulation and inference methods for non-Markovian stochastic biochemical reaction networks
School of Mathematical Sciences, Queensland University of Technology | Centre for Data Science, Queensland University of Technology | ARC Centre of Excellence for Mathematical Analysis of Cellular Systems (MACSYS), Queensland University of Technology
30秒速读
IN SHORT: This paper addresses the computational bottleneck of simulating and performing Bayesian inference for non-Markovian biochemical systems with history-dependent delays, which are crucial for modeling processes like gene transcription but are prohibitively expensive with existing methods.
核心创新
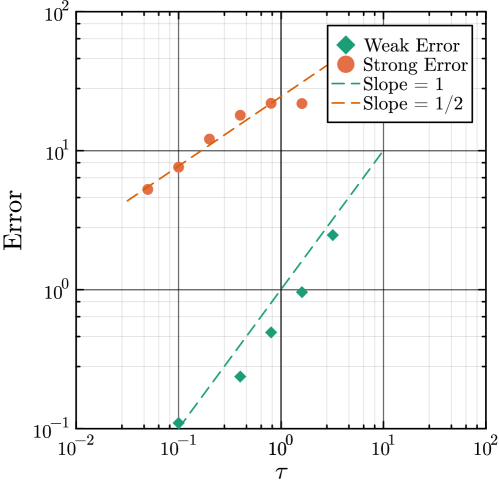
- Methodology Generalizes the next reaction method and τ-leaping to support arbitrary inter-event time distributions for non-Markovian systems, maintaining computational scalability.
- Methodology Introduces a novel coupling scheme to generate positively correlated exact and approximate non-Markovian sample paths, a prerequisite for variance reduction techniques.
- Methodology Enables the application of multifidelity and multilevel Monte Carlo (MLMC) methods to non-Markovian systems for the first time, bridging a significant methodological gap.
主要结论
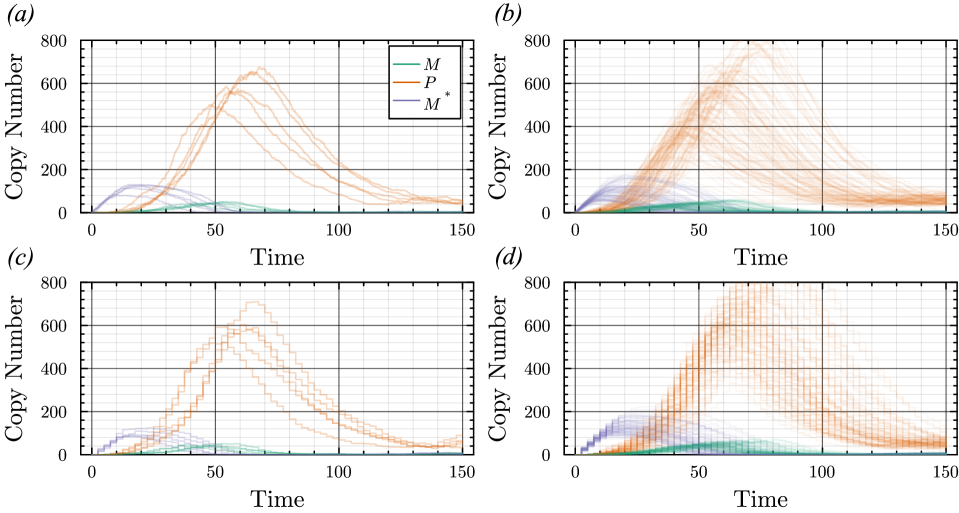
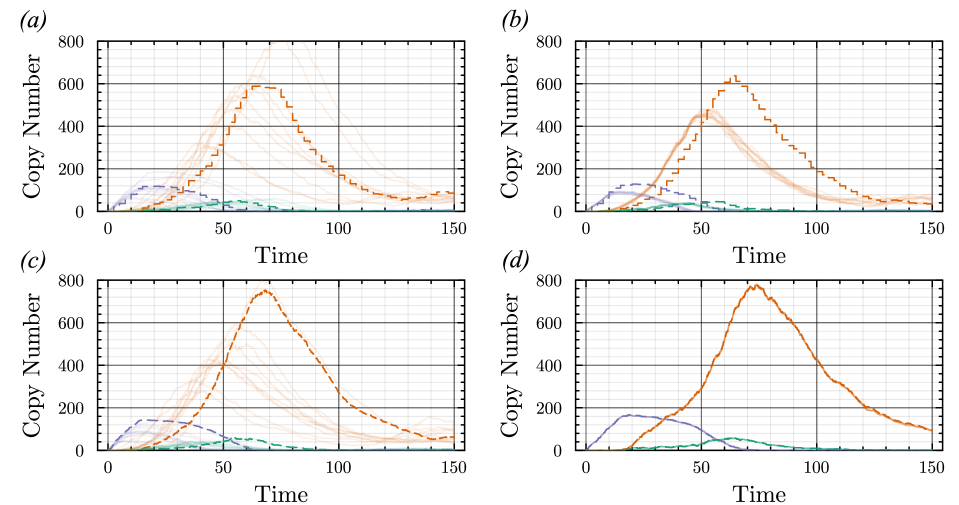
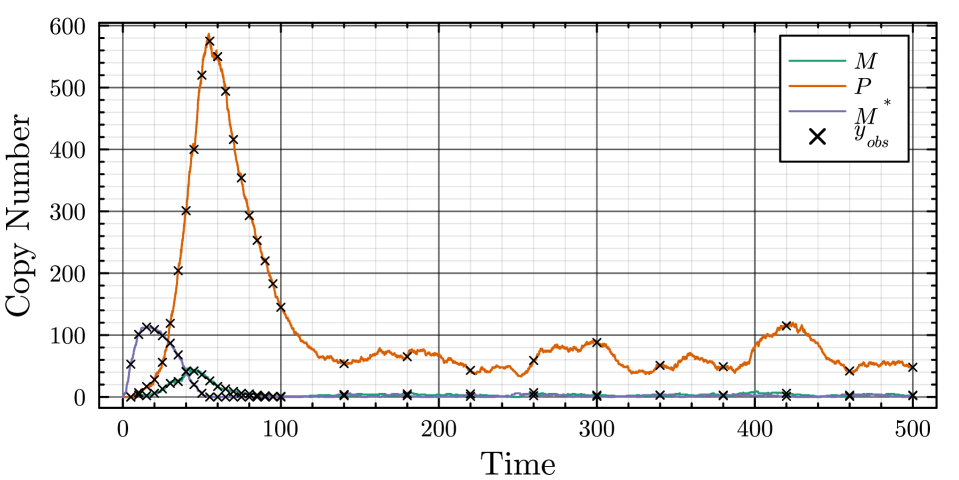
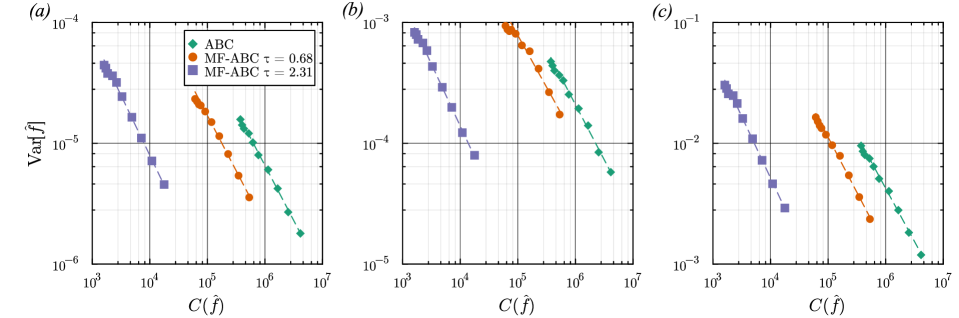
- The proposed non-Markovian simulation algorithms and coupling scheme successfully enable multifidelity inference, demonstrated on a gene regulation model with delayed auto-inhibition.
- The method achieves a computational speedup of two orders of magnitude (100x) in inference efficiency compared to standard approaches for the non-Markovian case study.
- The framework supports arbitrary delay distributions (state- and time-dependent), significantly extending the practical modeling scope beyond previous methods limited to simpler, time-only delays.
摘要: Stochastic models of biochemical reaction networks are widely used to capture intrinsic noise in cellular systems. The typical formulation of these models are based on Markov processes for which there is extensive research on efficient simulation and inference. However, there are biological processes, such as gene transcription and translation, that introduce history dependent dynamics requiring non-Markovian processes to accurately capture the stochastic dynamics of the system. This greater realism comes with additional computational challenges for simulation and parameter inference. We develop efficient stochastic simulation algorithms for well-mixed non-Markovian stochastic biochemical reaction networks with delays that depend on system state and time. Our methods generalize the next reaction method and τ-leaping method to support arbitrary inter-event time distributions while preserving computational scalability. We also introduce a coupling scheme to generate exact non-Markovian sample paths that are positively correlated to an approximate non-Markovian τ-leaping sample path. This enables substantial computational gains for Bayesian inference of model parameters though multifidelity simulation-based inference schemes. We demonstrate the effectiveness of our approach on a gene regulation model with delayed auto-inhibition, showing substantial gains in both simulation accuracy and inference efficiency of two orders of magnitude. These results extend the practical applicability of non-Markovian models in systems biology and beyond.