
Paper List
-
Discovery of a Hematopoietic Manifold in scGPT Yields a Method for Extracting Performant Algorithms from Biological Foundation Model Internals
This work addresses the core challenge of extracting reusable, interpretable, and high-performance biological algorithms from the opaque internal repr...
-
MS2MetGAN: Latent-space adversarial training for metabolite–spectrum matching in MS/MS database search
This paper addresses the critical bottleneck in metabolite identification: the generation of high-quality negative training samples that are structura...
-
Toward Robust, Reproducible, and Widely Accessible Intracranial Language Brain-Computer Interfaces: A Comprehensive Review of Neural Mechanisms, Hardware, Algorithms, Evaluation, Clinical Pathways and Future Directions
This review addresses the core challenge of fragmented and heterogeneous evidence that hinders the clinical translation of intracranial language BCIs,...
-
Less Is More in Chemotherapy of Breast Cancer
通过纳入细胞周期时滞和竞争项,解决了现有肿瘤-免疫模型的过度简化问题,以定量比较化疗方案。
-
Fold-CP: A Context Parallelism Framework for Biomolecular Modeling
This paper addresses the critical bottleneck of GPU memory limitations that restrict AlphaFold 3-like models to processing only a few thousand residue...
-
Open Biomedical Knowledge Graphs at Scale: Construction, Federation, and AI Agent Access with Samyama Graph Database
This paper addresses the core pain point of fragmented biomedical data by constructing and federating large-scale, open knowledge graphs to enable sea...
-
Predictive Analytics for Foot Ulcers Using Time-Series Temperature and Pressure Data
This paper addresses the critical need for continuous, real-time monitoring of diabetic foot health by developing an unsupervised anomaly detection fr...
-
Hypothesis-Based Particle Detection for Accurate Nanoparticle Counting and Digital Diagnostics
This paper addresses the core challenge of achieving accurate, interpretable, and training-free nanoparticle counting in digital diagnostic assays, wh...
On the Approximation of Phylogenetic Distance Functions by Artificial Neural Networks
Indiana University, Bloomington, IN 47405, USA
30秒速读
IN SHORT: This paper addresses the core challenge of developing computationally efficient and scalable neural network architectures that can learn accurate phylogenetic distance functions from simulated data, bridging the gap between simple distance methods and complex model-based inference.
核心创新
- Methodology Introduces minimal, permutation-invariant neural architectures (Sequence networks S and Pair networks P) specifically designed to approximate phylogenetic distance functions, ensuring invariance to taxa ordering without costly data augmentation.
- Methodology Leverages theoretical results from metric embedding (Bourgain's theorem, Johnson-Lindenstrauss Lemma) to inform network design, explicitly linking embedding dimension to the number of taxa for efficient representation.
- Methodology Demonstrates how equivariant layers and attention mechanisms can be structured to handle both i.i.d. and spatially correlated sequence data (e.g., models with indels or rate variation), adapting to the complexity of the generative evolutionary model.
主要结论
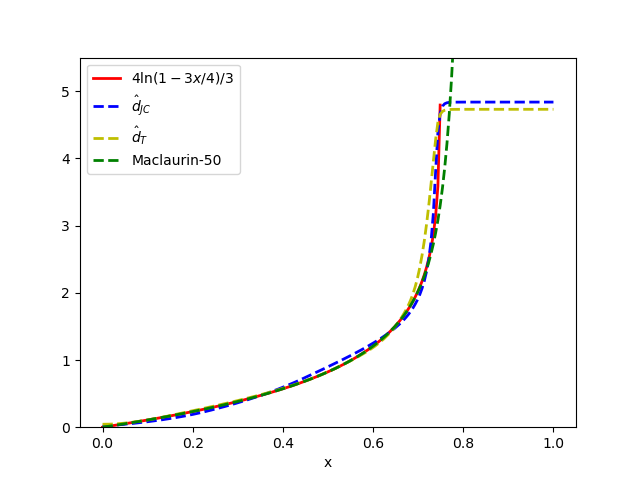
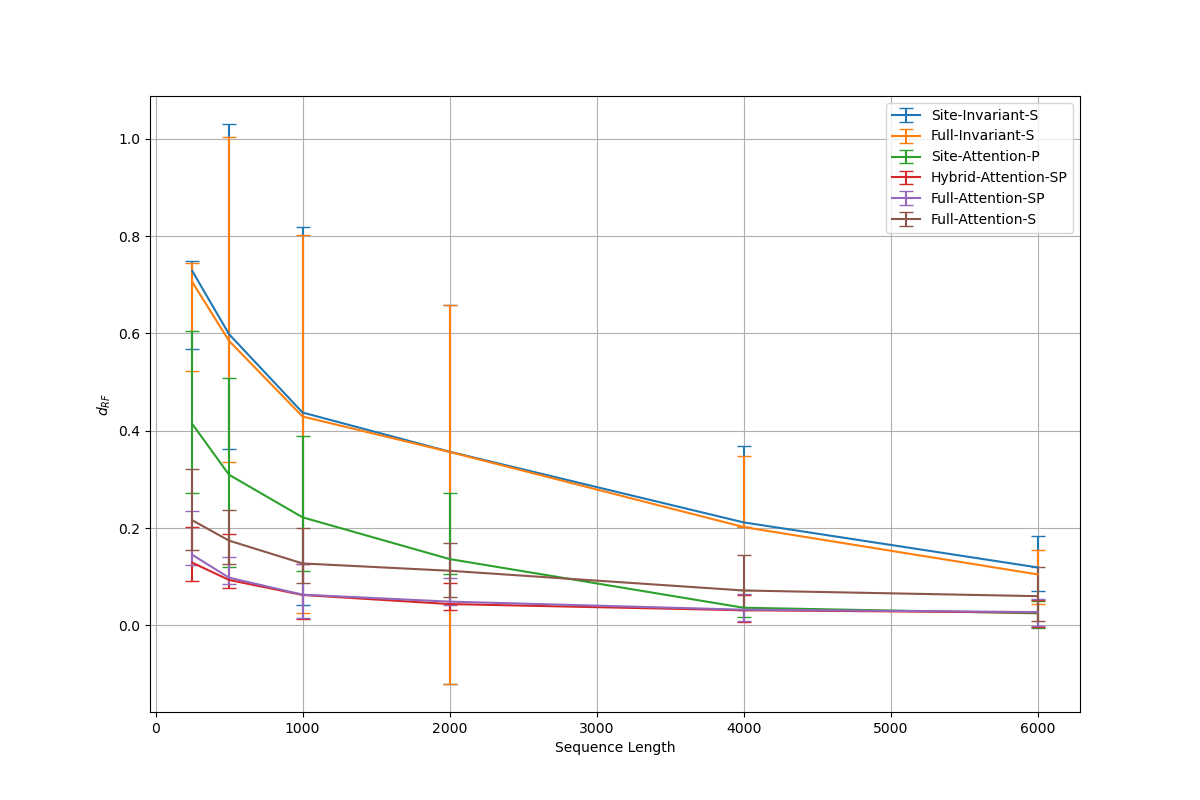
- The proposed minimal architectures (e.g., Sites-Invariant-S with ~7.6K parameters) achieve results comparable to state-of-the-art inference methods like IQ-TREE on simulated data under various models (JC, K2P, HKY, LG+indels), outperforming classic pairwise distance methods (d_H, d_JC, d_K2P) in most conditions.
- Architectures incorporating taxa-wise attention, while more memory-intensive, are necessary for complex evolutionary models with spatial dependencies; however, simpler networks suffice for simpler i.i.d. models, indicating an architecture-evolutionary model correspondence.
- Performance is highly sensitive to hyperparameters: validation error increases sharply with fewer than 4 attention heads or with hidden channel counts outside an optimal range (e.g., 32-128), aligning with theoretical requirements for learning graph-structured data.
摘要: Inferring the phylogenetic relationships among a sample of organisms is a fundamental problem in modern biology. While distance-based hierarchical clustering algorithms achieved early success on this task, these have been supplanted by Bayesian and maximum likelihood search procedures based on complex models of molecular evolution. In this work we describe minimal neural network architectures that can approximate classic phylogenetic distance functions and the properties required to learn distances under a variety of molecular evolutionary models. In contrast to model-based inference (and recently proposed model-free convolutional and transformer networks), these architectures have a small computational footprint and are scalable to large numbers of taxa and molecular characters. The learned distance functions generalize well and, given an appropriate training dataset, achieve results comparable to state-of-the art inference methods.