
Paper List
-
Discovery of a Hematopoietic Manifold in scGPT Yields a Method for Extracting Performant Algorithms from Biological Foundation Model Internals
This work addresses the core challenge of extracting reusable, interpretable, and high-performance biological algorithms from the opaque internal repr...
-
MS2MetGAN: Latent-space adversarial training for metabolite–spectrum matching in MS/MS database search
This paper addresses the critical bottleneck in metabolite identification: the generation of high-quality negative training samples that are structura...
-
Toward Robust, Reproducible, and Widely Accessible Intracranial Language Brain-Computer Interfaces: A Comprehensive Review of Neural Mechanisms, Hardware, Algorithms, Evaluation, Clinical Pathways and Future Directions
This review addresses the core challenge of fragmented and heterogeneous evidence that hinders the clinical translation of intracranial language BCIs,...
-
Less Is More in Chemotherapy of Breast Cancer
通过纳入细胞周期时滞和竞争项,解决了现有肿瘤-免疫模型的过度简化问题,以定量比较化疗方案。
-
Fold-CP: A Context Parallelism Framework for Biomolecular Modeling
This paper addresses the critical bottleneck of GPU memory limitations that restrict AlphaFold 3-like models to processing only a few thousand residue...
-
Open Biomedical Knowledge Graphs at Scale: Construction, Federation, and AI Agent Access with Samyama Graph Database
This paper addresses the core pain point of fragmented biomedical data by constructing and federating large-scale, open knowledge graphs to enable sea...
-
Predictive Analytics for Foot Ulcers Using Time-Series Temperature and Pressure Data
This paper addresses the critical need for continuous, real-time monitoring of diabetic foot health by developing an unsupervised anomaly detection fr...
-
Hypothesis-Based Particle Detection for Accurate Nanoparticle Counting and Digital Diagnostics
This paper addresses the core challenge of achieving accurate, interpretable, and training-free nanoparticle counting in digital diagnostic assays, wh...
Consistent Synthetic Sequences Unlock Structural Diversity in Fully Atomistic De Novo Protein Design
NVIDIA | Mila - Quebec AI Institute | Université de Montréal | HEC Montréal | CIFAR AI Chair
30秒速读
IN SHORT: This paper addresses the core pain point of low sequence-structure alignment in existing synthetic datasets (e.g., AFDB), which severely limits the performance of fully atomistic protein generative models.
核心创新
- Methodology Introduces a novel high-quality synthetic dataset (D_SYN-ours, ~0.46M samples) by leveraging ProteinMPNN for sequence generation and ESMFold for refolding, ensuring aligned and recoverable sequence-structure pairs.
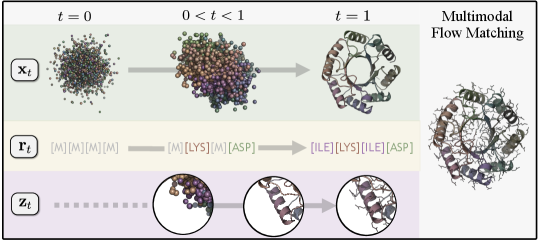
- Methodology Proposes Proteína-Atomística, a unified multi-modal flow-based framework that jointly models the distribution of Cα backbone atoms, discrete amino acid sequences, and non-Cα side-chain atoms in explicit observable space without latent variables.
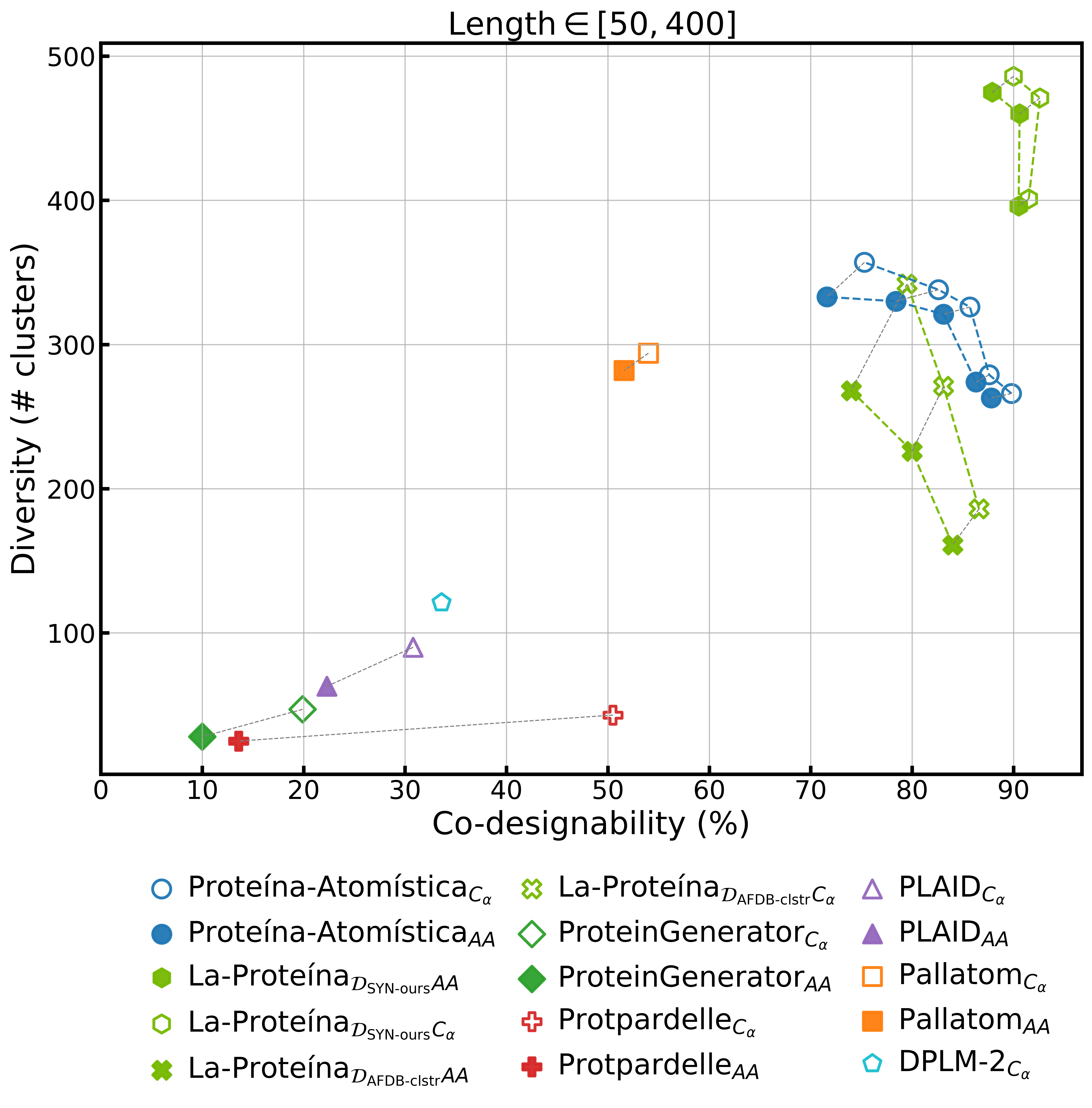
- Biology Demonstrates that consistent synthetic sequences are critical for unlocking structural diversity, with retrained La-Proteína achieving +54% structural diversity and +27% co-designability, and Proteína-Atomística achieving +73% structural diversity and +5% co-designability.
主要结论
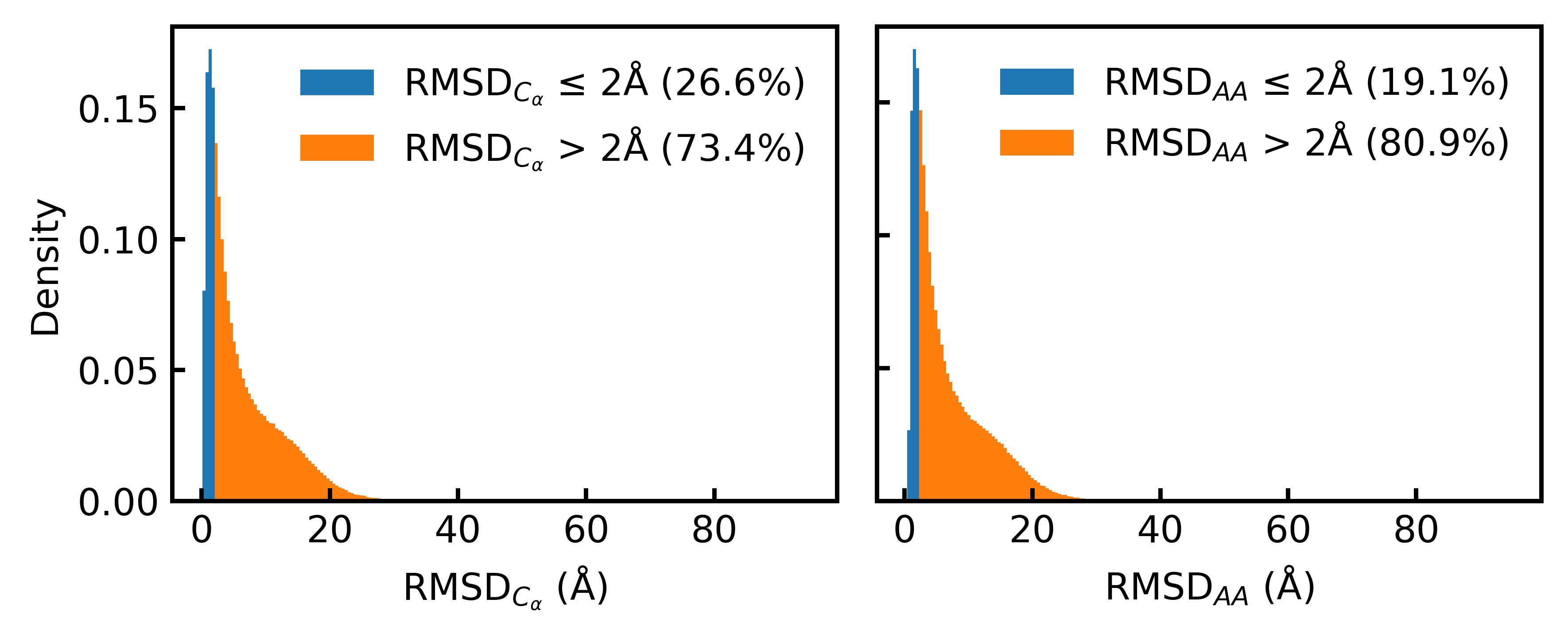
- Only 19.1% of the Foldseek-clustered AFDB dataset (D_AFDB-clstr) meets the standard 2Å all-atom RMSD co-designability threshold when refolded with ESMFold, revealing severe sequence-structure misalignment.
- Training on the new aligned dataset D_SYN-ours boosts La-Proteína's performance by +54% in structural diversity and +27% in co-designability, setting a new state-of-the-art.
- The proposed Proteína-Atomística framework, when trained on D_SYN-ours, shows a dramatic +73% improvement in structural diversity and a +5% improvement in co-designability, validating the dataset's broad utility.
摘要: High-quality training datasets are crucial for the development of effective protein design models, but existing synthetic datasets often include unfavorable sequence-structure pairs, impairing generative model performance. We leverage ProteinMPNN, whose sequences are experimentally favorable as well as amenable to folding, together with structure prediction models to align high-quality synthetic structures with recoverable synthetic sequences. In that way, we create a new dataset designed specifically for training expressive, fully atomistic protein generators. By retraining La-Proteína, which models discrete residue type and side chain structure in a continuous latent space, on this dataset, we achieve new state-of-the-art results, with improvements of +54% in structural diversity and +27% in co-designability. To validate the broad utility of our approach, we further introduce Proteína-Atomística, a unified flow-based framework that jointly learns the distribution of protein backbone structure, discrete sequences, and atomistic side chains without latent variables. We again find that training on our new sequence-structure data dramatically boosts benchmark performance, improving Proteína-Atomística’s structural diversity by +73% and co-designability by +5%. Our work highlights the critical importance of aligned sequence-structure data for training high-performance de novo protein design models. All data will be publicly released.