
Paper List
-
Nyxus: A Next Generation Image Feature Extraction Library for the Big Data and AI Era
This paper addresses the core pain point of efficiently extracting standardized, comparable features from massive (terabyte to petabyte-scale) biomedi...
-
Topological Enhancement of Protein Kinetic Stability
This work addresses the long-standing puzzle of why knotted proteins exist by demonstrating that deep knots provide a functional advantage through enh...
-
A Multi-Label Temporal Convolutional Framework for Transcription Factor Binding Characterization
This paper addresses the critical limitation of existing TF binding prediction methods that treat transcription factors as independent entities, faili...
-
Social Distancing Equilibria in Games under Conventional SI Dynamics
This paper solves the core problem of proving the existence and uniqueness of Nash equilibria in finite-duration SI epidemic games, showing they are a...
-
Binding Free Energies without Alchemy
This paper addresses the core bottleneck of computational expense in Absolute Binding Free Energy calculations by eliminating the need for numerous al...
-
SHREC: A Spectral Embedding-Based Approach for Ab-Initio Reconstruction of Helical Molecules
This paper addresses the core bottleneck in cryo-EM helical reconstruction: eliminating the dependency on accurate initial symmetry parameter estimati...
-
Budget-Sensitive Discovery Scoring: A Formally Verified Framework for Evaluating AI-Guided Scientific Selection
This paper addresses the critical gap in evaluating AI-guided scientific selection strategies under realistic budget constraints, where existing metri...
-
Probabilistic Joint and Individual Variation Explained (ProJIVE) for Data Integration
This paper addresses the core challenge of accurately decomposing shared (joint) and dataset-specific (individual) sources of variation in multi-modal...
Consistent Synthetic Sequences Unlock Structural Diversity in Fully Atomistic De Novo Protein Design
NVIDIA | Mila - Quebec AI Institute | Université de Montréal | HEC Montréal | CIFAR AI Chair
30秒速读
IN SHORT: This paper addresses the core pain point of low sequence-structure alignment in existing synthetic datasets (e.g., AFDB), which severely limits the performance of fully atomistic protein generative models.
核心创新
- Methodology Introduces a novel high-quality synthetic dataset (D_SYN-ours, ~0.46M samples) by leveraging ProteinMPNN for sequence generation and ESMFold for refolding, ensuring aligned and recoverable sequence-structure pairs.
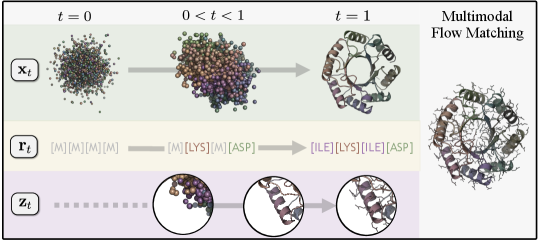
- Methodology Proposes Proteína-Atomística, a unified multi-modal flow-based framework that jointly models the distribution of Cα backbone atoms, discrete amino acid sequences, and non-Cα side-chain atoms in explicit observable space without latent variables.
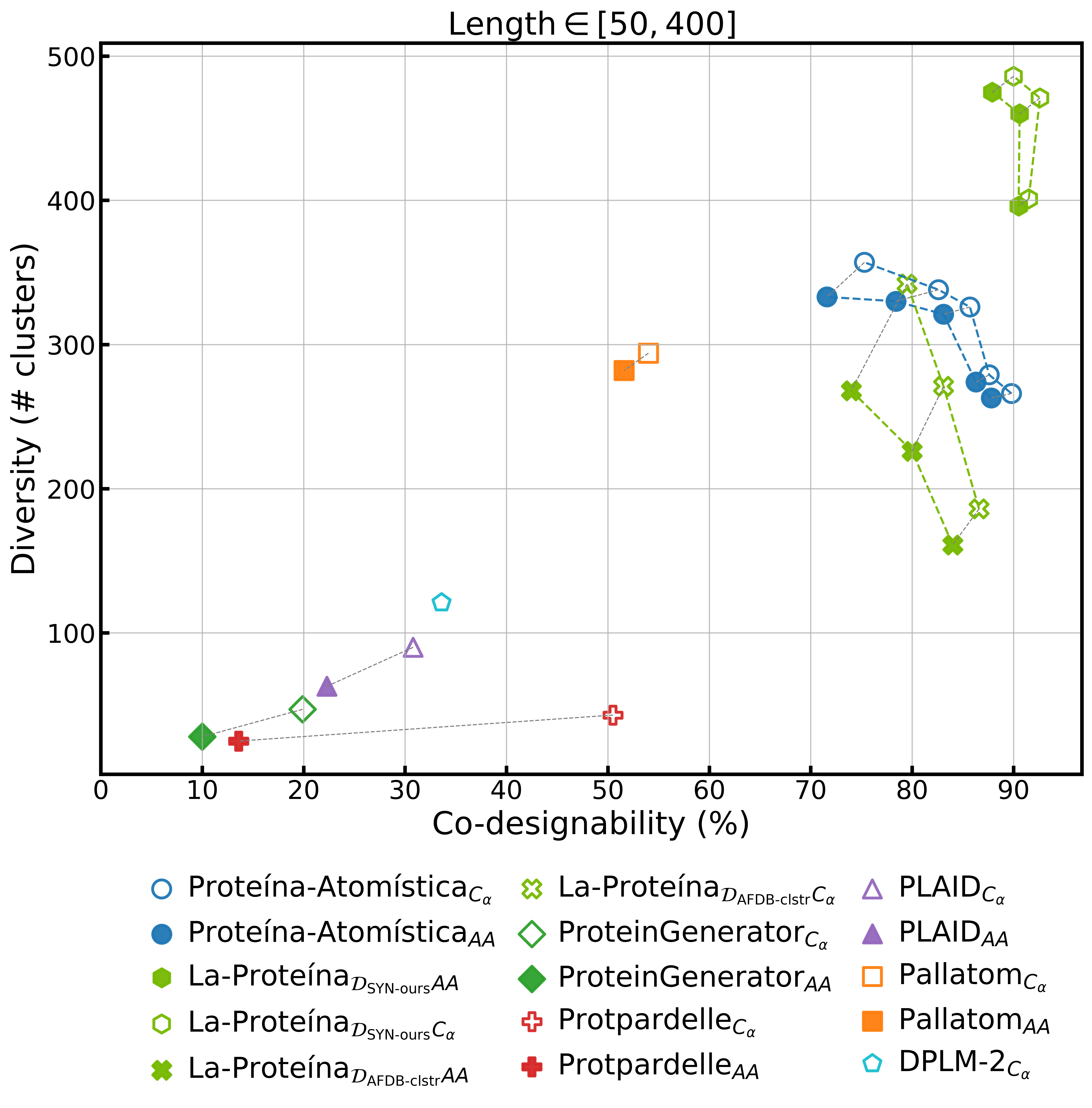
- Biology Demonstrates that consistent synthetic sequences are critical for unlocking structural diversity, with retrained La-Proteína achieving +54% structural diversity and +27% co-designability, and Proteína-Atomística achieving +73% structural diversity and +5% co-designability.
主要结论
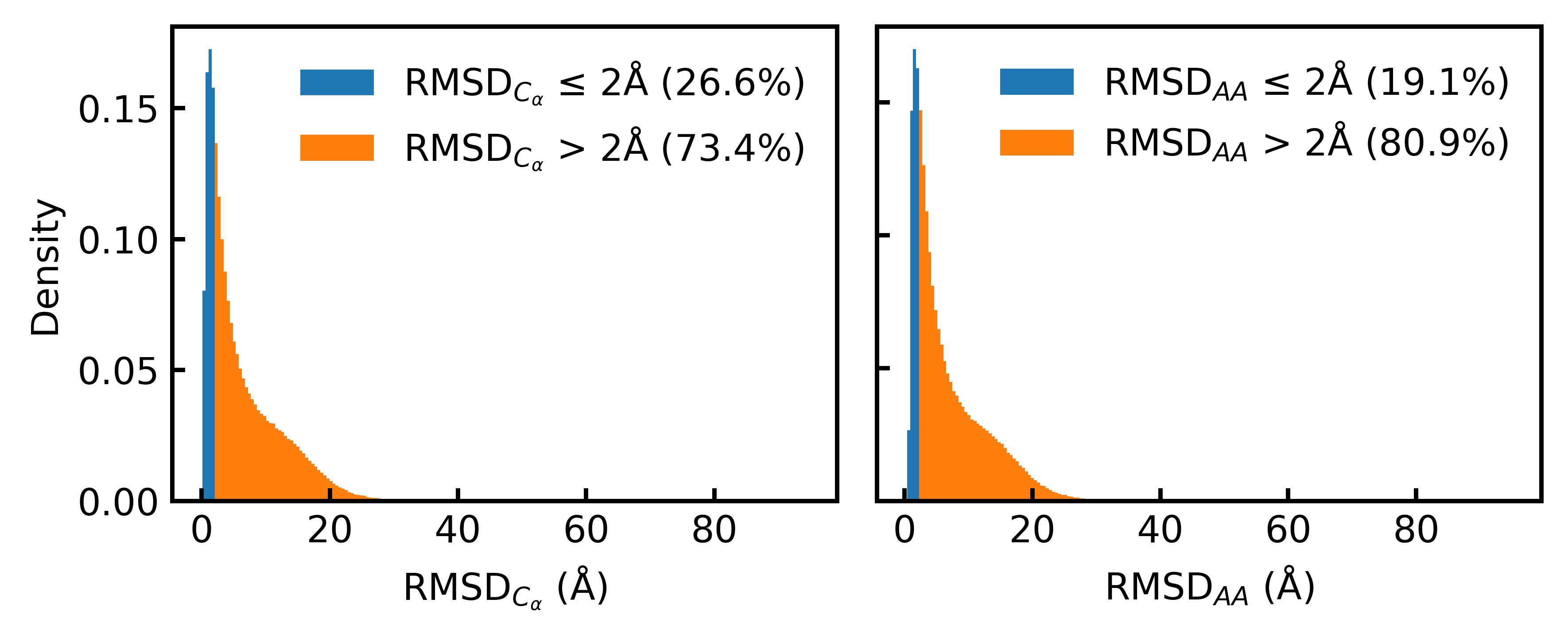
- Only 19.1% of the Foldseek-clustered AFDB dataset (D_AFDB-clstr) meets the standard 2Å all-atom RMSD co-designability threshold when refolded with ESMFold, revealing severe sequence-structure misalignment.
- Training on the new aligned dataset D_SYN-ours boosts La-Proteína's performance by +54% in structural diversity and +27% in co-designability, setting a new state-of-the-art.
- The proposed Proteína-Atomística framework, when trained on D_SYN-ours, shows a dramatic +73% improvement in structural diversity and a +5% improvement in co-designability, validating the dataset's broad utility.
摘要: High-quality training datasets are crucial for the development of effective protein design models, but existing synthetic datasets often include unfavorable sequence-structure pairs, impairing generative model performance. We leverage ProteinMPNN, whose sequences are experimentally favorable as well as amenable to folding, together with structure prediction models to align high-quality synthetic structures with recoverable synthetic sequences. In that way, we create a new dataset designed specifically for training expressive, fully atomistic protein generators. By retraining La-Proteína, which models discrete residue type and side chain structure in a continuous latent space, on this dataset, we achieve new state-of-the-art results, with improvements of +54% in structural diversity and +27% in co-designability. To validate the broad utility of our approach, we further introduce Proteína-Atomística, a unified flow-based framework that jointly learns the distribution of protein backbone structure, discrete sequences, and atomistic side chains without latent variables. We again find that training on our new sequence-structure data dramatically boosts benchmark performance, improving Proteína-Atomística’s structural diversity by +73% and co-designability by +5%. Our work highlights the critical importance of aligned sequence-structure data for training high-performance de novo protein design models. All data will be publicly released.