
Paper List
-
Formation of Artificial Neural Assemblies by Biologically Plausible Inhibition Mechanisms
This work addresses the core limitation of the Assembly Calculus model—its fixed-size, biologically implausible k-WTA selection process—by introducing...
-
How to make the most of your masked language model for protein engineering
This paper addresses the critical bottleneck of efficiently sampling high-quality, diverse protein sequences from Masked Language Models (MLMs) for pr...
-
Module control in youth symptom networks across COVID-19
This paper addresses the core challenge of distinguishing whether a prolonged societal stressor (COVID-19) fundamentally reorganizes the architecture ...
-
JEDI: Jointly Embedded Inference of Neural Dynamics
This paper addresses the core challenge of inferring context-dependent neural dynamics from noisy, high-dimensional recordings using a single unified ...
-
ATP Level and Phosphorylation Free Energy Regulate Trigger-Wave Speed and Critical Nucleus Size in Cellular Biochemical Systems
This work addresses the core challenge of quantitatively predicting how the cellular energy state (ATP level and phosphorylation free energy) governs ...
-
Packaging Jupyter notebooks as installable desktop apps using LabConstrictor
This paper addresses the core pain point of ensuring Jupyter notebook reproducibility and accessibility across different computing environments, parti...
-
SNPgen: Phenotype-Supervised Genotype Representation and Synthetic Data Generation via Latent Diffusion
This paper addresses the core challenge of generating privacy-preserving synthetic genotype data that maintains both statistical fidelity and downstre...
-
Continuous Diffusion Transformers for Designing Synthetic Regulatory Elements
This paper addresses the challenge of efficiently generating novel, cell-type-specific regulatory DNA sequences with high predicted activity while min...
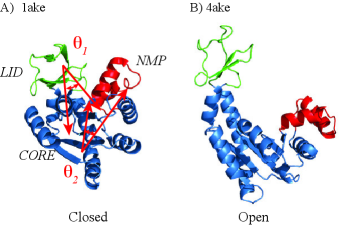
Realistic Transition Paths for Large Biomolecular Systems: A Langevin Bridge Approach
Department of Computer Science and Genome Center, University of California, Davis | Architecture et Dynamique des Macromolécules Biologiques, UMR 3528 du CNRS, Institut Pasteur | Department of Physics, School of Sciences, Great Bay University | Université Paris-Saclay, CNRS, CEA, Institut de Physique Théorique
30秒速读
IN SHORT: This paper addresses the core challenge of generating physically realistic and computationally efficient transition paths between distinct protein conformations, a problem where existing methods often produce non-physical trajectories due to oversimplified energy surfaces and steric clashes.
核心创新
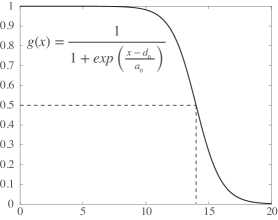
- Methodology Introduces SIDE (Stochastic Integro-Differential Equation), a novel Langevin bridge-based framework that efficiently approximates exact bridge equations at low temperatures to generate constrained transition trajectories.
- Methodology Develops a new coarse-grained potential that combines a Gō-like term (to preserve native backbone geometry) with a Rouse-type elastic energy term (from polymer physics), avoiding the problematic mixing of start/target conformation information used in prior methods like MinActionPath.
- Theory Provides a rigorous stochastic integro-differential formulation derived from the Langevin bridge formalism, which explicitly constrains trajectories to reach a target state within finite time, moving beyond Minimum Action Path (MAP) principles.
主要结论
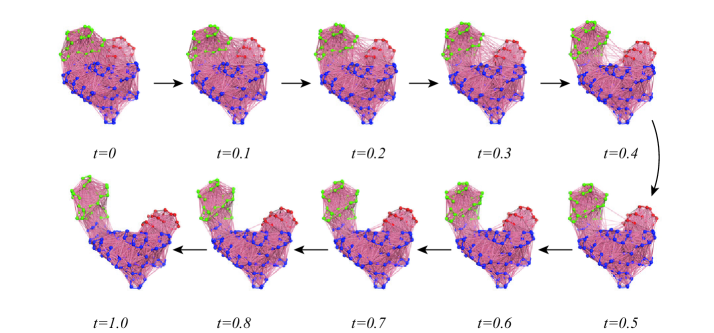
- The SIDE framework generates smooth, low-energy transition trajectories that maintain realistic molecular geometry, as demonstrated on several proteins undergoing large-scale conformational changes.
- SIDE frequently recovers experimentally supported intermediate states along transition paths, suggesting its paths have biological relevance beyond mere endpoint interpolation.
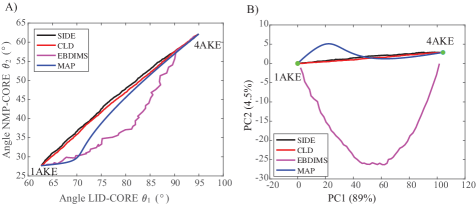
- Compared to established methods like MinActionPath and EBDIMS, SIDE offers improved physical realism and computational efficiency for modeling biomolecular conformational transitions, though challenges remain for highly complex motions.
摘要: We introduce a computational framework for generating realistic transition paths between distinct conformations of large biomolecular systems. The method is built on a stochastic integro-differential formulation derived from the Langevin bridge formalism, which constrains molecular trajectories to reach a prescribed final state within a finite time and yields an efficient low-temperature approximation of the exact bridge equation. To obtain physically meaningful protein transitions, we couple this formulation to a new coarse-grained potential combining a Gō-like term that preserves native backbone geometry with a Rouse-type elastic energy term from polymer physics; we refer to the resulting approach as SIDE. We evaluate SIDE on several proteins undergoing large-scale conformational changes and compare its performance with established methods such as MinActionPath and EBDIMS. SIDE generates smooth, low-energy trajectories that maintain molecular geometry and frequently recover experimentally supported intermediate states. Although challenges remain for highly complex motions—largely due to the simplified coarse-grained potential—our results demonstrate that SIDE offers a powerful and computationally efficient strategy for modeling biomolecular conformational transitions.